
03 August 2022
Neural networks allows ab initio simulation of water viscosity


The viscosity of fluids is a crucial parameter playing a fundamental role in many fields of science and technology, ranging from chemical and mechanical engineering, to biochemical processes and planetary sciences, to name but a few. The viscosity can be computed from equilibrium molecular dynamics using the Green-Kubo (GK) theory of linear response, which relates the viscosity ( or in general, the transport coefficients) to correlation functions of the out-of-diagonal element of the stress tensor (or the corresponding flux). Despite its apparent simplicity, the application of GK for the viscosity hides few challenges, arising mainly from the noisy nature of the correlation functions that enter the GK relations. For this reason, long simulation times are required to achieve acceptable statistical accuracy, thus despite the great abundance of work on the properties of water from ab initio simulation, its viscous properties have been dodged since it was well established in the literature that “an accurate computation of the viscosity of water would require exceedingly long first-principles simulations” [1].
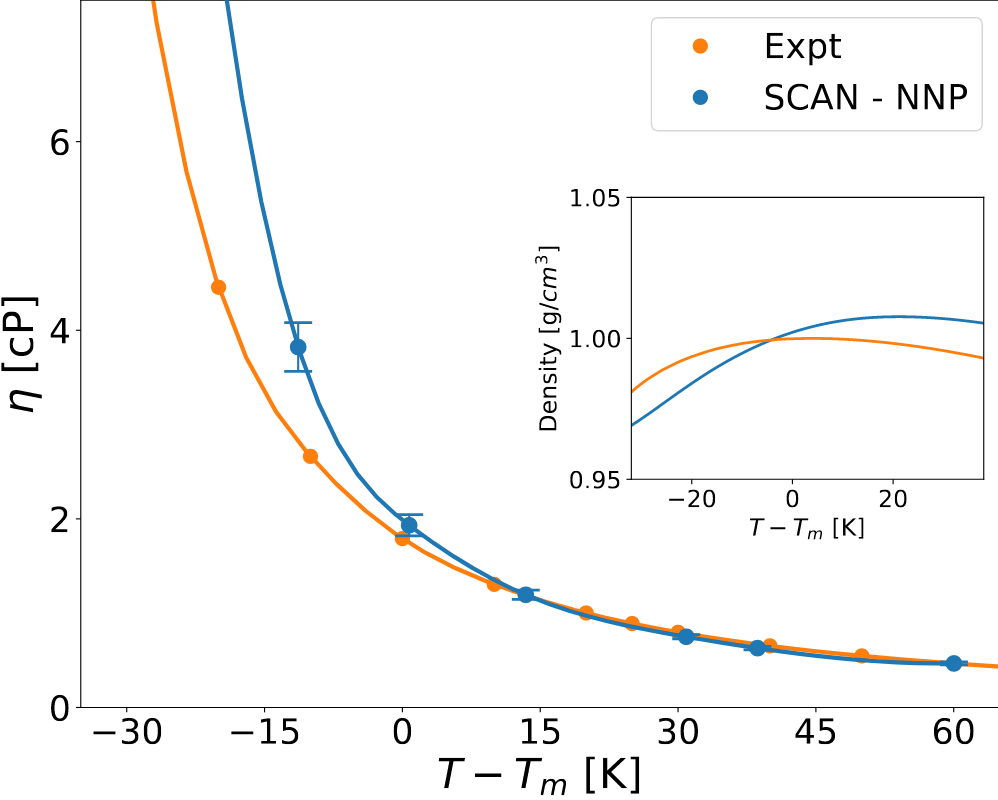
In our work to cope with the long simulation times necessary to achieve acceptable statistical accuracy, our ab initio approach is enhanced with deep-neural-network potentials (NNP). This approach is first validated against AIMD results, obtained by using the Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional and paying careful attention to crucial, yet often overlooked, aspects of the statistical data analysis. Then, we train a second NNP to a dataset generated from the Strongly Constrained and Appropriately Normed (SCAN) functional. Once the error resulting from the imperfect prediction of the melting line is offset by referring the simulated temperature to the theoretical melting one, our SCAN predictions of the shear viscosity of water are in excellent agreement with experiments.
The work is part of a collaboration between a group in SISSA (Stefano Baroni, Cesare Malosso and Davide Tisi) and a group in Princeton ( Roberto Car and Linfeng Zhang).
The group led by Stefano Baroni has a great experience in the computation of transport properties of materials, and, recently, it has developed a method to accurately and efficiently compute them using the cepstral analysis [2].
The transport properties require long simulation times that limit the possibility of an extensive ab initio exploration of the phase diagram. To solve this problem, we used the neural network potential developed at Princeton by Linfeng Zhang and Roberto Car [3].
The two groups have also recently collaborated on the computation of thermal conductivity via ab initio and NN potentials [4].
Finally, we report on an extensive study of the viscosity of liquid water at near-ambient conditions, performed within the Green-Kubo theory of linear response and equilibrium ab initio molecular dynamics (AIMD), based on density-functional theory (DFT). In order to cope with the long simulation times necessary to achieve acceptable statistical accuracy, our ab initio approach is enhanced with deep-neural-network potentials (NNP). This approach is first validated against AIMD results, obtained by using the Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional and paying careful attention to crucial, yet often overlooked, aspects of the statistical data analysis. Then, we train a second NNP to a dataset generated from the Strongly Constrained and Appropriately Normed (SCAN) functional. Our study confirms the ability of the SCAN exchange-correlation density functional to predict a broad array of properties of water over a wide range of thermodynamic conditions. Minor shortcomings observed in the undercooled regime are possibly related to the subtle balance between the high- and low-density fluctuations that become more prominent upon undercooling as one approaches the hypothesized metastable liquid-liquid critical point.
[1] LaCount, M. D. & Gygi, F. Ensemble first-principles molecular dynamics simulations of water using the SCAN meta-GGA density functional. The J. Chem.Phys. 151,164101, DOI: https://doi.org/10.1063/1.5124957 (2019)
[2] Ercole, L., Marcolongo, A. & Baroni, S. Accurate thermal conductivities from optimally short molecular dynamics simulations. Sci. Reports 7, 15835, DOI: https://doi.org/10.1038/s41598-017-15843-2 (2017).
[3] Zhang, L. et al. End-to-end symmetry preserving inter-atomic potential energy model for finite and extended systems. In Bengio, S. et al. (eds.) Advances in Neural Information Processing Systems 31, 4436–4446 (Curran Associates, Inc., 2018).
[4] D Tisi, L Zhang, R Bertossa, H Wang, R Car, S Baroni, Heat transport in liquid water from first-principles and deep neural network simulations, Physical Review B 104 (22), 224202, DOI: 10.1103/PhysRevB.104.224202